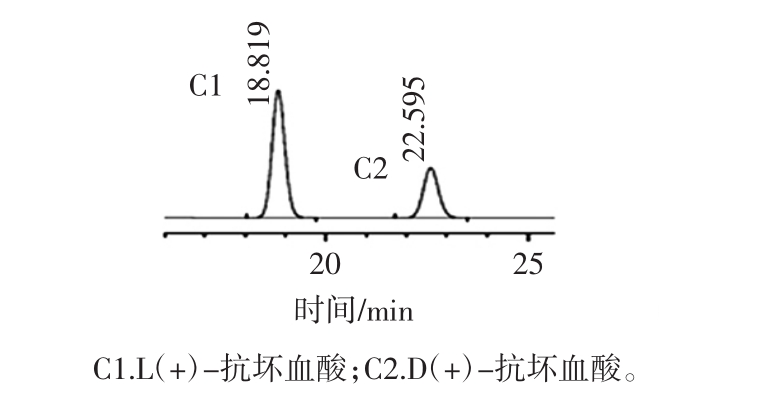
图1 对照品色谱图
Fig.1 Chromatogram of reference substance
刘成浩,张蓉,邬国庆,杜勇,杨玲,刘齐,韩萧茜,张华珺 *
(北京市药品检验所,北京102200)
摘 要: 采用高效液相色谱法,建立一种可同时测定保健食品中抗坏血酸的两种同分异构体(L(+)-抗坏血酸和D(+)-抗坏血酸)含量的方法,并分析保健食品原料中抗坏血酸的组成成分以及L(+)-抗坏血酸的稳定性。采用资生堂CAPCELL PAK C18 TYPE-AQ色谱柱,以磷酸二氢钾溶液-甲醇(98∶2)为流动相,流速1.0 mL/min,检测波长为245 nm,进样量20 μL。结果显示,在优化的条件下,抗坏血酸的两种异构体分离度为2.51,能够排除杂质干扰,进行准确定量,L(+)-抗坏血酸和 D(+)-抗坏血酸的浓度线性范围分别为 2 μg/mL~100 μg/mL(r 2 =0.999 7)和 2 μg/mL~100 μg/mL(r 2 =0.999 9),平均回收率分别为 100.2%和 101.6%,检出限均为 1.3 mg/100 g。
关键词: 抗坏血酸;高效液相色谱法;稳定性;同分异构体;含量
抗坏血酸有两种同分异构体,分别为L(+)-抗坏血酸和D(+)-抗坏血酸。L(+)-抗坏血酸具有强还原性,对人体具有生物活性,可以防治坏血病,并且在过敏性皮肤病、肝炎等疾病上也有治疗作用。但L(+)-抗坏血酸不能在人体内合成,需要从外界摄取,因此广泛应用于保健食品中。另外L(+)-抗坏血酸极不稳定,遇空气中氧、热、碱等会被氧化成脱氢L(+)-抗坏血酸,被氧化后的L(+)-抗坏血酸在人体内活性大大降低;D(+)-抗坏血酸又称为异抗坏血酸,虽然抗氧化性强于L(+)-抗坏血酸,但是对人体基本无生物活性,摄入过多会引起尿酸结石、多尿、皮疹等 [1-10] ,因此,国家食品药品监督管理总局发布的《营养素补充剂原料目录》(2016年第205号公告)中明确规定D(+)-抗坏血酸不能用于保健食品中。
目前测定抗坏血酸含量的方法中荧光法及滴定法应用较广泛,然而荧光法操作繁琐,稳定性较差;滴定法易受样品颜色干扰,误差较大 [11-17] 。并且上述方法只能测定样品中天然存在或添加的抗坏血酸总量,无法分别出保健食品原料添加的是L(+)-抗坏血酸还是D(+)-抗坏血酸,由于国家规定 D(+)-抗坏血酸不能用于保健食品中,因此应用上述方法均不能对保健食品市场进行有效监管,保健食品原料添加情况存在一定风险。2016年8月31日颁布实施的《食品安全国家标准-食品中抗坏血酸的测定》(GB 5009.86-2016) [18] 对于抗坏血酸含量测定新增了高效液相色谱法,此方法可以同时测定食品中抗坏血酸两种同分异构体L(+)-抗坏血酸和D(+)-抗坏血酸。但是,日常监管检测中发现标准给出的实验条件应用于保健食品时不能很好的分离两种同分异构体并且操作步骤复杂,故本文探讨了不同色谱条件下两种同分异构体的分离情况,对方法进行了优化,发现采用资生堂CAPCELL PAK C18 TYPE-AQ耐水柱,以磷酸二氢钾溶液-甲醇(98∶2)为流动相,无需控制pH即可较好分离L(+)-抗坏血酸和D(+)-抗坏血酸并分别准确测定其含量,方法简便重复性好。优化后的方法可以很好地测定保健食品中抗坏血酸的含量并检测保健食品厂家原料中是否违规添加D(+)-抗坏血酸,为日常监管消除了风险。同时此方法还可分析保健食品中L(+)-抗坏血酸的稳定性 [19-22] ,通过此方法考察保健食品不同剂型L(+)-抗坏血酸稳定性可以为企业改进生产方法作为参考。
高效液相色谱仪(1260 Infinity):Agilent;电子天平(XS205):Mettle。
甲醇(色谱纯):德国默克。磷酸二氢钾、L-半胱氨酸(均为优级纯)、磷酸、偏磷酸、磷酸三钠(均为分析纯):国药集团。十六烷基三甲基溴化铵(色谱纯):SIGMA公司。
L(+)-抗坏血酸对照品:(来源:Dr.Ehrenstorfer GmbH 纯度:98.9%批号:103944)。D(+)-抗坏血酸对照品(来源:TRC 纯度:98%批号:1-AGE-94-1)。
1.2.1 标准品溶液制备
精密称取 L(+)-抗坏血酸对照品 10 mg和 D(+)-抗坏血酸对照品10 mg,置于100 mL棕色容量瓶中,用20 g/L偏磷酸溶液溶解并稀释至刻度。精密量取上述对照品储备液 0.2、0.5、1.0、2.5、5.0、10.0 mL 分别置于10 mL棕色容量瓶中,加20 g/L偏磷酸溶液稀释至刻度,摇匀备用。
1.2.2 供试品溶液的制备
在避光条件下,取本品20片研细混匀,精密称取0.2 g,置于50 mL棕色容量瓶中,用20 g/L偏磷酸溶液溶解并定容,摇匀后过滤,弃去初滤液,从续滤液中精密吸取2.0 mL,置于50 mL棕色容量瓶中,用20 g/L偏磷酸溶液稀释定容至刻度,混匀。0.45 μm微孔滤膜滤过,滤液上机测定L(+)-抗坏血酸和D(+)-抗坏血酸的含量。
1.2.3 供试品中脱氢抗坏血酸的还原
从1.2.2供试品溶液中精密吸取2.0 mL,置于50 mL离心管中,用20 g/L偏磷酸溶液补至20 mL,加入40 g/L的L-半胱氨酸溶液10 mL,用100 g/L磷酸三钠溶液调节pH值至7.1,以200次/min振荡5 min。再用磷酸调节pH值至4.5,用水将试液全部转移至50 mL容量瓶中,并定容至刻度,混匀。0.45 μm微孔滤膜滤过,滤液上机测定L(+)-抗坏血酸总量,其中包含抗坏血酸和脱氢抗坏血酸,并由此可推算出抗坏血酸氧化程度。
1.2.4 色谱条件
色谱柱:资生堂CAPCELL PAK C18 TYPE-AQ柱,柱长 250 mm,内径 4.6 mm,粒径 5 μm;流动相:磷酸二氢钾溶液(取13.600 g磷酸二氢钾和1.821 g十六烷基三甲基溴化铵,加水溶解并定容至2 000 mL,用 0.45 μm 微孔滤膜滤过即得)-甲醇(98∶2);流速:1.0 mL/min;检测波长为 245 nm;进样量 20 μL。理论板数应大于2 000,各峰分离度应大于2。
2.1.1 色谱柱的选择
本试验中选用3根不同的色谱柱,分别为资生堂CAPCELLPAKC18TYPE-AQ、安捷伦ZORBAXSB-Aq、德国MN C18柱,按1.2.4中色谱条件分别进行试验,考察不同色谱柱对色谱分离的影响,其色谱图见图1。
图1 对照品色谱图
Fig.1 Chromatogram of reference substance
结果表明资生堂CAPCELL PAK C18 TYPE-AQ色谱柱分离度较好,不同色谱柱的分离度见表1。
表1 色谱柱对色谱分离的影响
Table 1 The influence of chromatogram by column
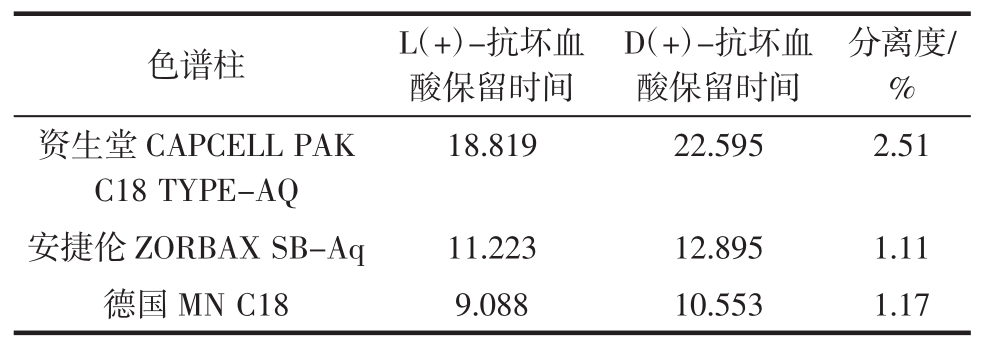
表1表明,资生堂CAPCELL PAK C18 TYPE-AQ色谱柱分离效果最好(色谱图见图1),因此采用资生堂CAPCELL PAK C18 TYPE-AQ为本实验色谱柱。
2.1.2 流动相的pH值对色谱分离的影响
以磷酸二氢钾溶液-甲醇(98∶2)为流动相,改变其pH值,考察pH值对L(+)-抗坏血酸和D(+)-抗坏血酸色谱行为的影响。结果见表2。
表2 pH对色谱分离的影响
Table 2 The influence of chromatogram by pH

表2表明,流动相pH值为4.0、4.5、5.0时分离度均满足要求,且当流动相按上述方法配制后pH值为4.5,在不改变pH值的条件下即可很好的分开抗坏血酸两种同分异构体。
2.2.1 线性关系
取 1.2.1 中 L(+)-抗坏血酸和 D(+)-抗坏血酸标准品溶液,在1.2.4色谱条件下用高效液相色谱仪测定,记录色谱图,经统计学处理得线性回归方程L(+)-抗坏血酸:A=45.09 C-83.71,相关系数 r=0.999 7;D(+)-抗坏血酸:A=43.26C-64.30,相关系数 r=0.999 9。表明两种抗坏血酸同分异构体在2 μg/mL~100 μg/mL浓度范围内,峰面积与浓度均呈良好的线性关系。
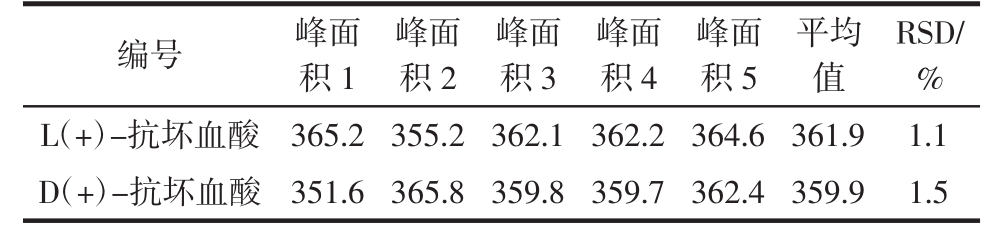
2.2.2 精密度实验
取1.2.1标准曲线中第3点(10 μg/mL浓度)重复进样5次,记录色谱图,计算RSD值为L(+)-抗坏血酸:1.1%;D(+)-抗坏血酸 1.5%。
表3 精密度测定结果
Table 3 The results of precision
2.2.3 加标回收实验
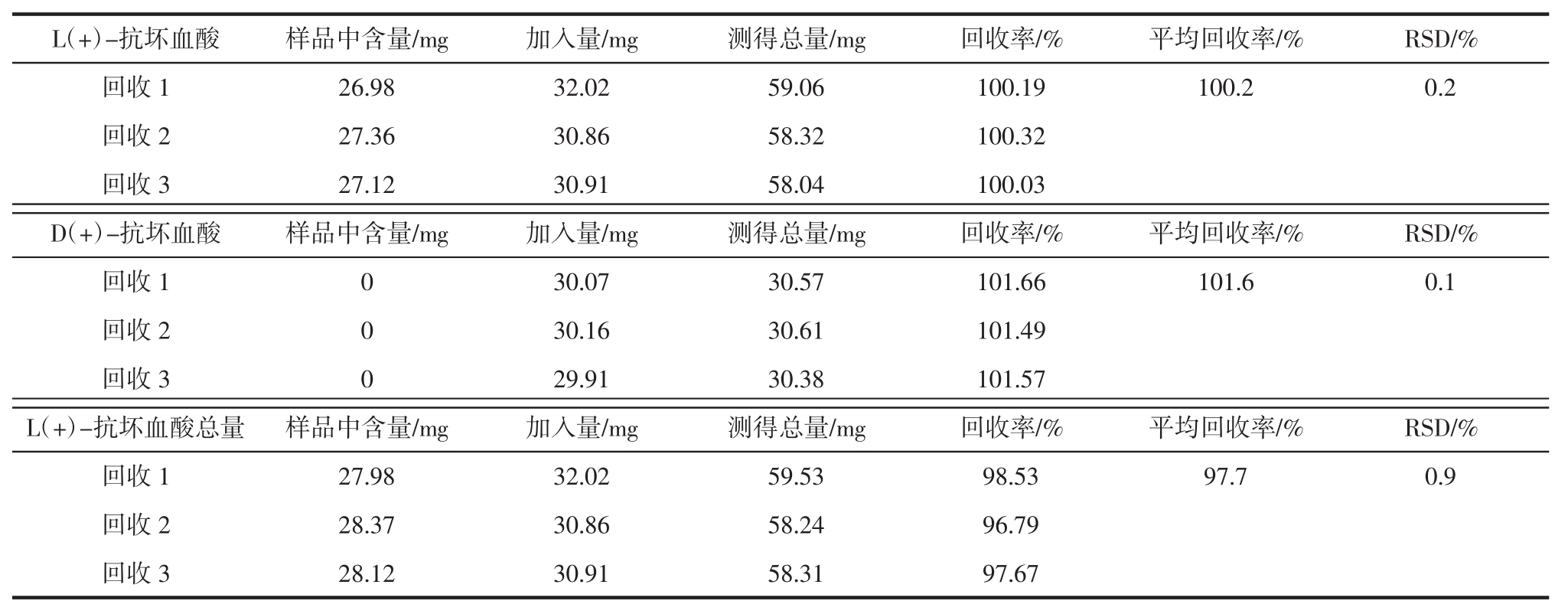
取已知含量的供试品(含量为L(+)-抗坏血酸:2.69×10 4 mg/100 g;D(+)-抗坏血酸:0 mg/100 g;按1.2.3测定 L(+)-抗坏血酸总量为 2.79×10 4 mg/100 g)研细混匀后,精密称取 0.100 3、0.101 7、0.100 8 g,分别置于50 mL棕色容量瓶中,精密称取L(+)-抗坏血酸和D(+)-抗坏血酸对照品各30 mg,置于上述50 mL棕色容量瓶中,用20 g/L偏磷酸溶液溶解并定容,摇匀后过滤,弃去初滤液,从续滤液中精密吸取2.0 mL,置于50 mL棕色容量瓶中,用20 g/L偏磷酸溶液稀释定容至刻度,混匀。0.45 μm微孔滤膜滤过,滤液上机测定L(+)-抗坏血酸和D(+)-抗坏血酸的加标回收率。
从上述续滤液中精密吸取2.0 mL,置于50 mL离心管中,用20 g/L偏磷酸溶液补至20 mL,加入40 g/L的L-半胱氨酸溶液10 mL,用100 g/L磷酸三钠溶液调节pH至7.1,以200次/min振荡5 min。再用磷酸调节pH至4.5,用水将试液全部转移至50 mL容量瓶中,并定容至刻度,混匀。0.45 μm微孔滤膜滤过,滤液上机测定L(+)-抗坏血酸总量的加标回收率。加标回收率结果见表4。
表4 回收率结果
Table 4 The results of recoveries
2.3.1 对于市售保健食品中抗坏血酸组成的分析
由于保健食品营养素补充剂原料中不包括D(+)-抗坏血酸,因此随机抽取市售保健食品中销量较大的30个厂家的品种,在本文条件下进行试验,考察其中抗坏血酸成分的组成,发现均未检出D(+)-抗坏血酸,因此可认为这30批次样品均为合格产品。
2.3.2 保健食品中L(+)-抗坏血酸稳定性的分析
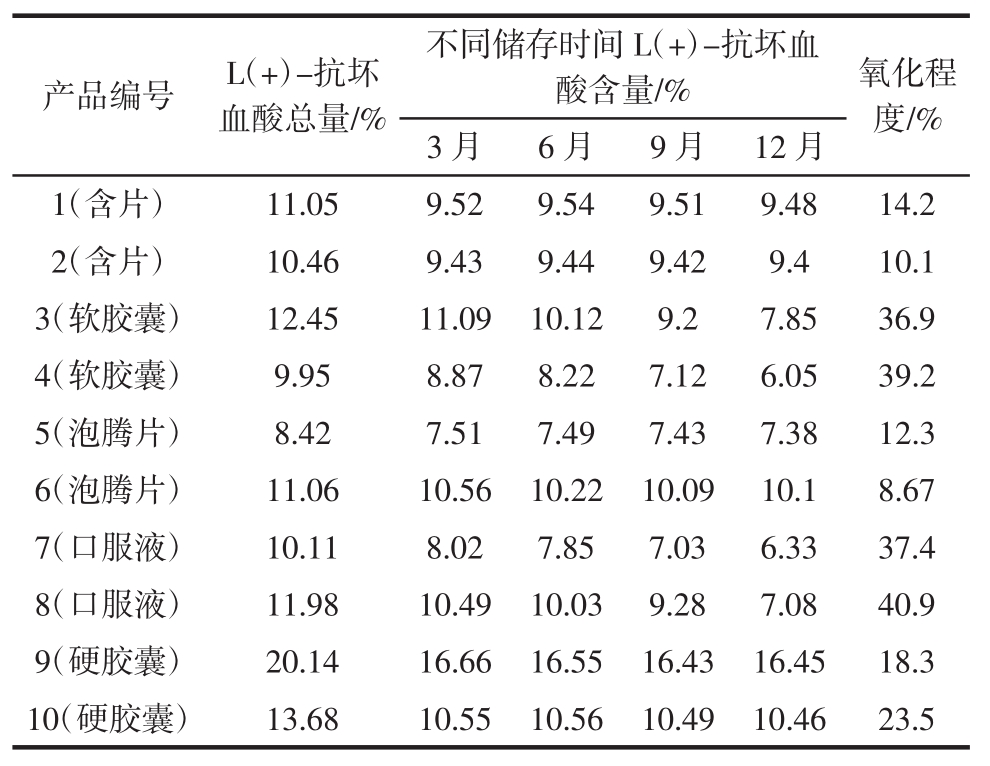
随机抽取市售保健食品不同剂型的10批次产品进行稳定性试验,在温度为25℃,相对湿度为50%的条件下放置12个月,分别在3、6、9、12个月时取样测定其L(+)-抗坏血酸的含量,取0月的样品进行1.2.3中供试品还原的操作,测定L(+)-抗坏血酸总量,以此比较出保健食品中抗坏血酸的氧化程度。结果见表5。
表5表明,大部分储存 3、6、9、12月后的产品L(+)-抗坏血酸氧化程度较小,而软胶囊和口服液中抗坏血酸含量随时间延长含量有所降低,氧化程度均接近40%,由此推测在适宜的储存环境下固体产品稳定性较好,液态产品稳定性稍差。
表5 L(+)-抗坏血酸稳定性的分析
Table 5 The analysis of stability
1)本试验通过考察不同pH值对测定结果以及L(+)-抗坏血酸和D(+)-抗坏血酸分离度的影响,发现国标中给出的pH2.5~2.8条件下分离度不能满足R≥1.5,而按照本文1.2.4中条件配制流动相时,其pH值为4.5不需要再进行调节pH值的步骤即可满足分离度R≥1.5,简化了国标方法的同时优化了试验条件。
2)本试验中考察了3根不同的色谱柱,分别为资生堂CAPCELL PAK C18 TYPE-AQ、安捷伦ZORBAX SB-Aq、德国MN C18柱。结果表明资生堂CAPCELL PAK C18 TYPE-AQ柱耐水性较好,分离效果最好,故采用此色谱柱作为本试验的色谱柱。
3)由于抗坏血酸极易氧化,所以试验中用水均为新沸晾凉后的水,并且在避光条件下进行试验,以减少抗坏血酸氧化对结果的影响。通过本试验方法可以对保健食品中抗坏血酸的稳定性进行研究,采用1.2.3中方法将样品进行还原测定其L(+)-抗坏血酸总量,与直接测定的L(+)-抗坏血酸含量进行比较从而判断产品中抗坏血酸的被氧化程度。研究中发现目前大部分保健食品在正常储存环境下抗坏血酸较稳定,个别产品中L(+)-抗坏血酸被氧化程度较高,其中液态产品稳定性较固态产品差,可能由于产品包装透光性较强,密封不严以及液态环境下更有利于抗坏血酸分解所致,因此建议厂家可改用片剂或泡腾片等剂型。
4)目前,国家标准中抗坏血酸的含量测定有滴定法,荧光法及HPLC法3种,荧光法操作繁琐,稳定性较差;滴定法易受样品颜色干扰,误差较大;新增的HPLC法相较于前两种方法优势在于稳定性好重复性高,并且可以分离 L(+)-抗坏血酸和D(+)-抗坏血酸两种异构体,由此可应用于保健食品中抗坏血酸组成的测定,从而监督保健食品厂家是否添加了营养素补充剂原料中不允许添加的D(+)-抗坏血酸。与此同时,此方法还可以测得L(+)-抗坏血酸被氧化程度,而被氧化的L(+)-抗坏血酸对人体活性较小,可以利用此方法研究L(+)-抗坏血酸在保健食品的不同制备及存储条件下的稳定性从而选择最优方案,使产品质量控制得到更好的提升。
参考文献:
[1]杨媛,冯晓元,石磊,等.高效液相色谱法同时测定水果蔬菜中L-抗坏血酸、D-异抗坏血酸、脱氢抗坏血酸及总维生素C的含量[J].分析测试学报,2015,34(8):934-938
[2]张冬梅,汪振立,罗六保,等.对新鲜果蔬中维生素C的测定结果影响因素研究[J].江西化工,2010(1):73-76
[3]丁健桦,饶火瑜,王兴祥.HPLC法初步研究维生素C的稳定性[J].食品工业,2004,25(1):44-45
[4]李荣玮,何晓艳,潘振宇.HPLC法测定小儿复方赖氨酸颗粒中维生素C的含量[J].中国药师,2016,19(8):1588-1589
[5]谢巍,李玉静.湿热环境对维C银翘片中维生素C含量的影响[J].中国卫生检验杂志,2017,27(5):628-631
[6]郑培,张庆云,袁丽云,等.医药中抗坏血酸检测方法研究[J].医学研究与教育,2015,32(2):65-68
[7]刘根,孙登明.银掺杂聚L-精氨酸修饰电极同时测定芦丁和抗坏血酸[J].分析科学学报,2014,30(1):83-86
[8]张进,何鑫,姚思童,等.漫反射傅里叶变换红外光谱法测定维生素C制剂中抗坏血酸的含量[J].化学分析计量,2013,22(5):51-54
[9]刘育坚,王丹,许志刚.HILIC色谱柱拆分抗坏血酸对映体及其在药物分析中的应用[J].化学研究,2017,28(6):726-729
[10]陈静,魏晓亮,陈雅琴.高效液相色谱法测定多维矿物质片中维生素C的含量[J].辽宁化工,2017,46(10):1039-1041
[11]WEN Y P,DUAN X M,XU J K,et al.One-step electrosynthesis of poly(3,4-ethylenedioxy-thiophene)-ethylsulfate matrix for fabricating vitamin C electrochemical biosensor and its determination in commercial juices[J].Chinese Journal of Polymer Science,2012,30(3):460-469
[12]Hiroshi Iwase.Routine high-performance liquid chromatographic determination of ascorbic acid in foods using L-methionine for the pre-analysis sample stabilization[J].Talanta,2003,60(5):1011-1021
[13]张秀芹,王敏,顾莹,等.HPLC检测食品中L-抗坏血酸、D-异抗坏血酸及抗坏血酸总量[J].中国卫生检验杂志,2013,23(9):2056-2061
[14]郝学宁,郝嫱嫱,刘雪莲.HPLC法测定果蔬中维生素C[J].现代农业科技,2011,3(3):30-31
[15]张清,李刚.高压液相色谱法同时测定食品中抗坏血酸与异抗坏血酸的含量[J].食品安全质量检测学报,2013,4(1):251-256
[16]姚立国,许大陆,郑洁,等.碘量法和分光光度法测定萝卜中维生素C含量的比较研究[J].山东化工,2015,44(13):74-76
[17]李野,尹利辉,高尚,等.食品和药品中维生素C含量测定方法的研究进展[J].药物分析杂志,2016,36(5):756-764
[18]中华人民共和国国家卫生和计划生育委员会.GB 5009.86-2016食品安全国家标准食品中抗坏血酸的测定-液相色谱法[S].北京:中国标准出版社
[19]王敬,刘彩虹,马育松,等.高效液相色谱法测定强化食品中维生素C的含量[J].化学分析计量,2014,23(6):29-33
[20]关皓月,杨锐,孙会敏.HPLC法测定L-抗坏血酸中的有关物质及探讨[J].中国药事,2015,29(11):1184-1188
[21]P H M Marfil,E M Santos,V R N Telis,et al.Ascorbic Acid Degradation Kinetics in Tomatoes at Different Drying Conditions[J].Food Science and Technology,2008,41(9):1642-1647
[22]陈雪,吴林,李刚,等.食品中总抗坏血酸和总异抗坏血酸HPLC法测定[J].中国公共卫生,2011,27(2):160-161
LIU Cheng-hao,ZHANG Rong,WU Guo-qing,DU Yong,YANG Ling,LIU Qi,HAN Xiao-qian,ZHANG Hua-jun *
(Beijing Institute for Drug Control,Beijing 102200,China)
Abstract: A high performance liquid chromatography method was developed for simultaneous determination of two isomers which were L(+)-ascorbic acid and D(+)-ascorbic acid in health food,and the degree of oxidation of L(+)-ascorbic acid could be analyzed by this method.The separation was performed on a Shiseido CAPCELL PAK C18 TYPE-AQ column.The mobile phase consisted of potassium dihydrogen phosphate solution and methanol(98 ∶2).The flow rate was 1.0 mL/min.The detection UV wavelength was at 245 nm.The injection volume was 20 μL.The results showed that by this method,the two isomers could be quantitative analyzed and the degree of separation was 2.51,the linear ranges of L(+)-ascorbic acid and D(+)-ascorbic acid were 2 μg/mL-100 μg/mL(r 2 =0.999 7)and 2 μg/mL-100 μg/mL(r 2 =0.999 9),The spiked recoveries of the two analytes were 100.2%and 101.6%,The limits of detection(LOD)were 1.3 mg/100 g.
Key words: ascorbic acid;high performance liquid chromatography(HPLC);stability;isomeride;content
The Analysis of Composition and Stability of Ascorbic Acid in Health Food by HPLC
LIU Chenghao,ZHANG Rong,WU Guoqing,et al.The Analysis of Composition and Stability of Ascorbic Acid in Health Food by HPLC[J].Food Research and Development,2018,39(18):172-176
刘成浩,张蓉,邬国庆,等.HPLC法测定保健食品中抗坏血酸的组成及稳定性[J].食品研究与开发,2018,39(18):172-176
*通信作者: 张华珺(1981—),女(汉),副主任药师,硕士。
引文格式:
作者简介: 刘成浩(1992—),男(满),药师,学士,研究方向:保健食品化妆品检验。
收稿日期: 2018-03-13
DOI: 10.3969/j.issn.1005-6521.2018.18.031